题目:WDR45 contributes to neurodegeneration through regulation of ER homeostasis and neuronal death
期刊:Autophagy
影响因子:11.05
主要技术: CRISPAR-Cas9、WB、IHC、TMT蛋白组学 、PRM蛋白靶向验证
研究背景
宏自噬是降解受损细胞器和蛋白质聚集体的主要细胞分解代谢过程。在人类中,在自噬的多个步骤中起作用的各种基因的突变可引起广泛的神经退行性疾病。小鼠WDR45神经元特异性敲除可导致自噬和轴突变性功能障碍,运动协调性差和学习记忆障碍。因此,WDR45的主要功能是调节自噬体的形成,其功能的丧失会导致小鼠行为异常。但WDR45的丧失如何导致BPAN中的神经变性的机制仍不清楚。为了解决这些问题,本文作者构建了WDR45敲除小鼠来模拟BPAN病变的复杂过程,并且利用TMT定量蛋白质组学技术和一系列细胞水平的验证实验来阐明BPAN样病理变化的分子机制。
技术路线
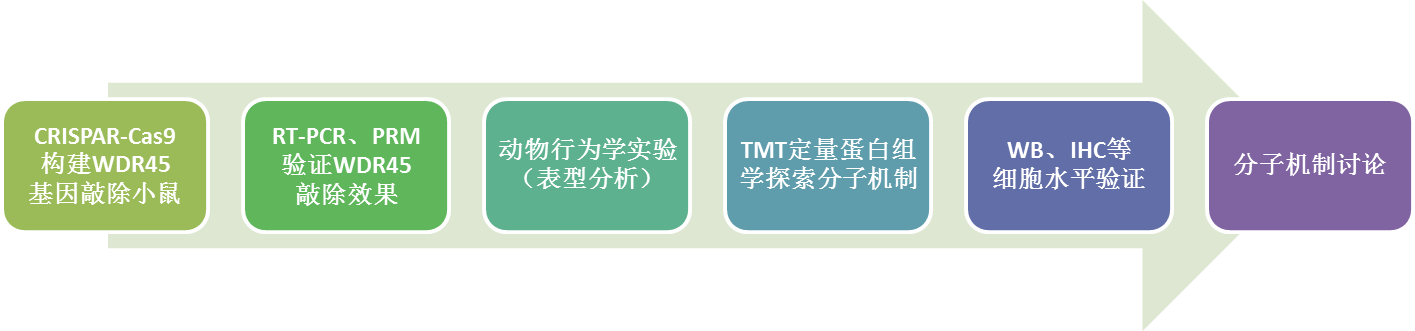
实验结果
1.WDR45基因敲除小鼠显示认知障碍
首先作者利用CRISPAR-Cas9技术成功构建了WDR45基因敲除小鼠。
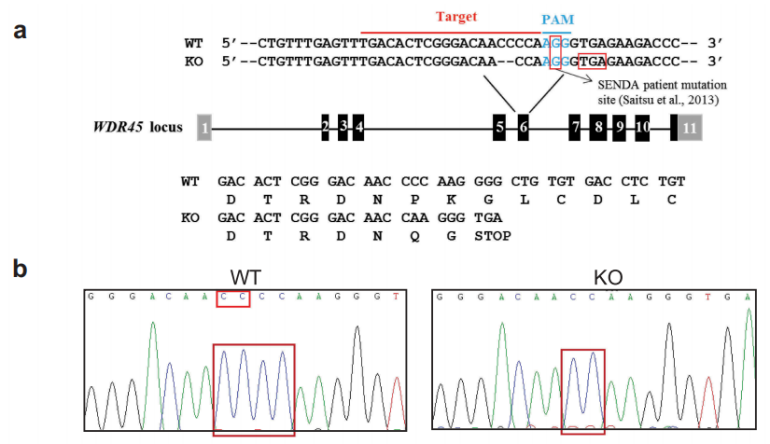
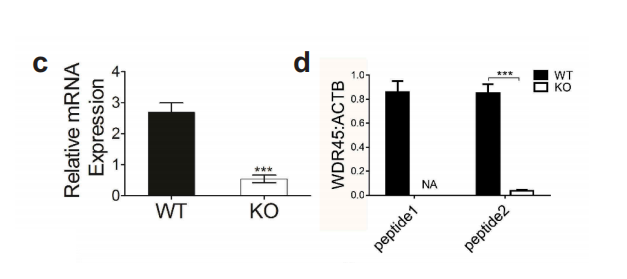
图1-1 成功构建WDR45基因敲除小鼠
接下来作者利用6月龄野生型小鼠和WDR45基因敲除小鼠进行了一系列的行为学实验,包括morris水迷宫实验、8臂迷宫试验、条件性恐惧实验、旋转实验、三箱社交实验、Pilocarpine诱发癫痫实验等。结果表明实验月龄的WDR45 敲除小鼠在运动协调方面没有明显损害的迹象,但是在空间学习和条件记忆方面存在认知障碍。另外,与野生型小鼠相比,KO小鼠表现出明显地更短的潜伏期和更严重的癫痫发作。
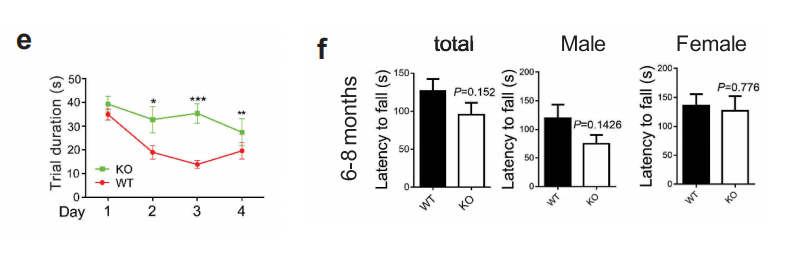
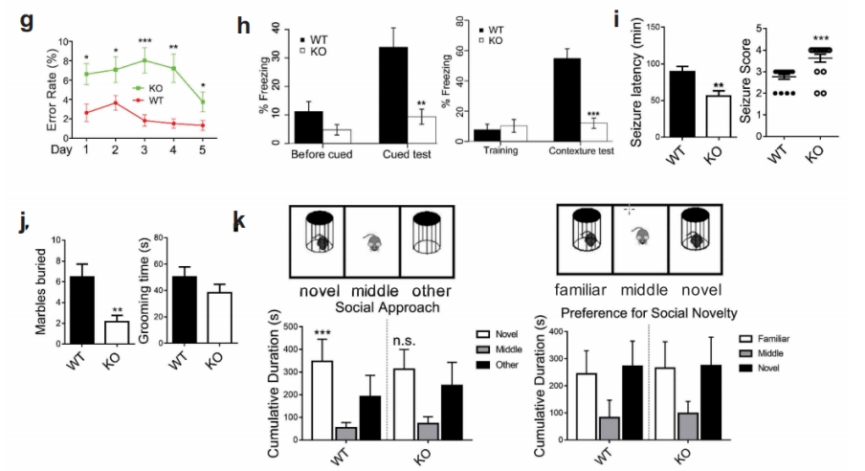
图1 行为学实验,显示WDR45基因敲除小鼠具有认知障碍
2.WDR45基因敲除小鼠脑内出现神经元丢失
由于BPAN患者成年后表现为神经变性病和帕金森病,因此作者利用免疫组化(IHC)和TUNEL染色等方法检查了老年小鼠是否有神经变性的迹象。结果表明:WDR45敲除小鼠在老年时在前额叶皮质和黑质中表现出神经元丢失。
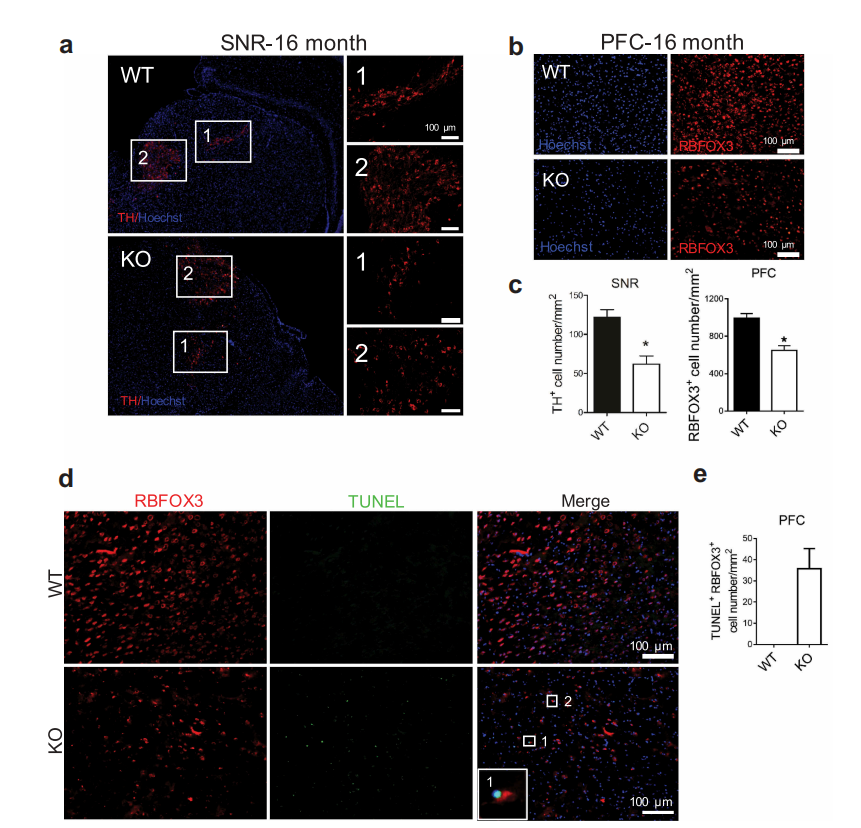
图2 WDR45敲除小鼠在老年时在前额叶皮质和黑质中表现出神经元丢失
3.脑组织定量蛋白质组学分析揭示了内质网(ER)蛋白在基因敲除小鼠脑中的积累
作者设计了3个10标TMT定量蛋白组学实验,分别研究了5对野生型(WT)小鼠和敲除(KO)小鼠的大脑前额叶皮层(PFC)、海马(HIP)和中脑(MIB)组织的差异定量蛋白组学。共定量到近4000个蛋白,以FC>1.5,P<0.05的标准,在PFC、HIP和HIP组织中分别筛选到67、12、185个显著差异蛋白,其中PFC和MIB共有30个显著差异蛋白,HIP和MIB共有4个显著差异蛋白。定量结果的主成分分析表明,两种基因型在不同脑区的蛋白质表达谱是完全分离的。这些结果说明利用质谱定量的方法能够通过WDR45的缺失来区分小鼠脑组织蛋白质组。

图3 WT和KO小鼠脑组织蛋白质组定量分析
另外,生信分析结果表明,内质网(ER)蛋白在基因敲除小鼠脑中积累最为显著,其次是脂质代谢过程和内膜系统组织。
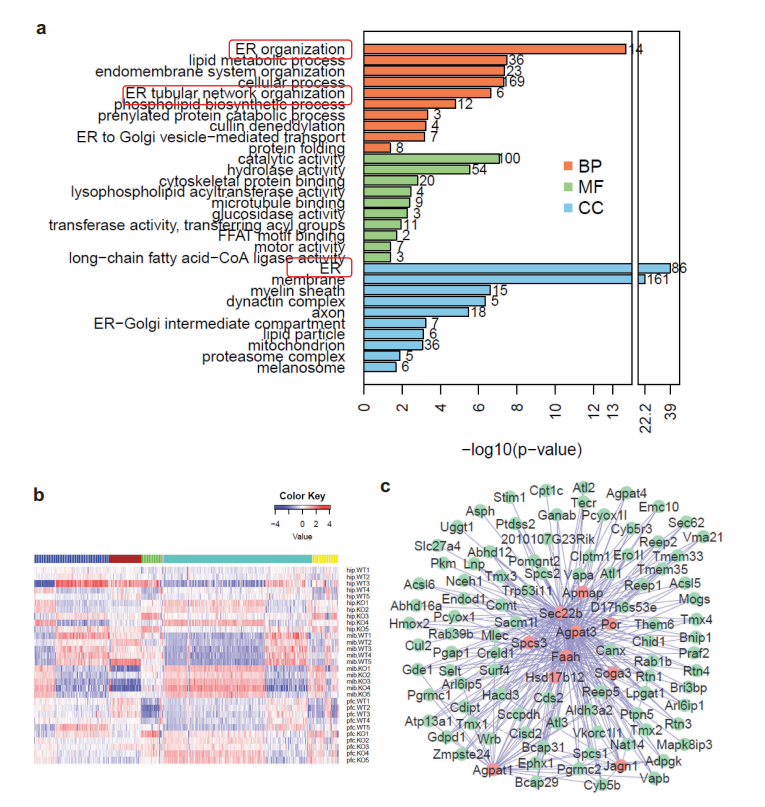
图4 生物信息学分析显示内质网(ER)蛋白在WDR45 KO小鼠体内积累
4.WDR45通过蛋白酶体和溶酶体途径调节靶ER蛋白
接下来,作者研究WDR45如何调节内质网蛋白的水平。首先,Western blot 实验结果证实WDR45 KO小鼠中脑内源性SEC22B显著上调,与质谱定量结果趋势一致。然后,作者在存在或缺乏BAFA1(一种阻止自噬体溶酶体融合的自噬抑制剂)或MG132(一种蛋白酶体抑制剂)的情况下共表达WDR45和4个内质网蛋白,结果表明,BAFA1可有效地阻断WDR45介导的SEC22B降解,而BAFA1和 MG132均可阻断WDR45介导的BCAP31降解。而对照组,只有MG132对GFP的稳定性有轻微影响。这些不同的作用结果表明,位于内质网不同部位的蛋白质被不同的机制降解。此外,在细胞中通过shRNA敲低WDR45,与敲低Scrbl 相比,所有四种ER蛋白质在sh-WDR45的细胞都有积累,与KO小鼠脑中观察到的效果类似。这些结果表明WDR45对内质网蛋白亚群的稳定性有负面影响。
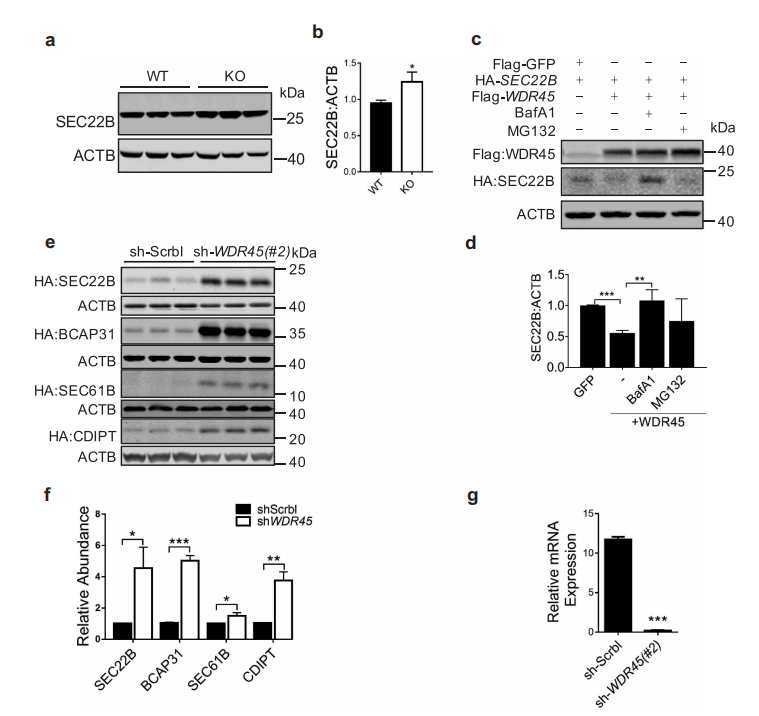
图5 WDR45介导靶标内质网(ER)蛋白的降解
5.WDR45缺失细胞可导致ER扩增,增加ER应激
随后,作者研究了ER蛋白的积累是否与ER面积的扩大相关。通过标记内源性ER蛋白CALR(calreticulin),shRNA敲低WDR45与与敲低sh-SCRBL的细胞相比,显示出明显的内质网扩张。此外,使用衣霉素诱导内质网应激可导致外源WDR45被聚集到WT的ER中,而在WDR45缺失细胞没有这种现象。免疫荧光分析结果表明,在内质网应激后,在WT皮质神经元的溶酶体(红色)中积累了CALR蛋白(绿色),表明内质网蛋白的溶酶体降解需要WDR45。为了验证内质网自噬的这种作用是否是细胞器特异性,作者用羰基*化物间氯苯腙(CCCP)处理细胞来破坏线粒体膜电位。对照组细胞经CCCP处理24小时后,PRKN染色明显减少,线粒体明显萎缩,提示PRKN介导了细胞的有丝分裂。然而,在敲低表达sh-WDR45的细胞中,线粒体明显增大。最后,对小鼠黑质的透射电镜分析显示KO小鼠内质网小管增大。因此,作者证明了宏自噬的缺陷导致了内质网和线粒体这两个独立细胞器的自噬缺陷。
此外,作者发现,诱导内质网应激可进一步促进ER伴侣蛋白HSPA5的表达。在KO小鼠的原代神经元中,作者还发现HSPA5表达增加,ERN1/IRE1激活,XBP1选择性剪接增加。
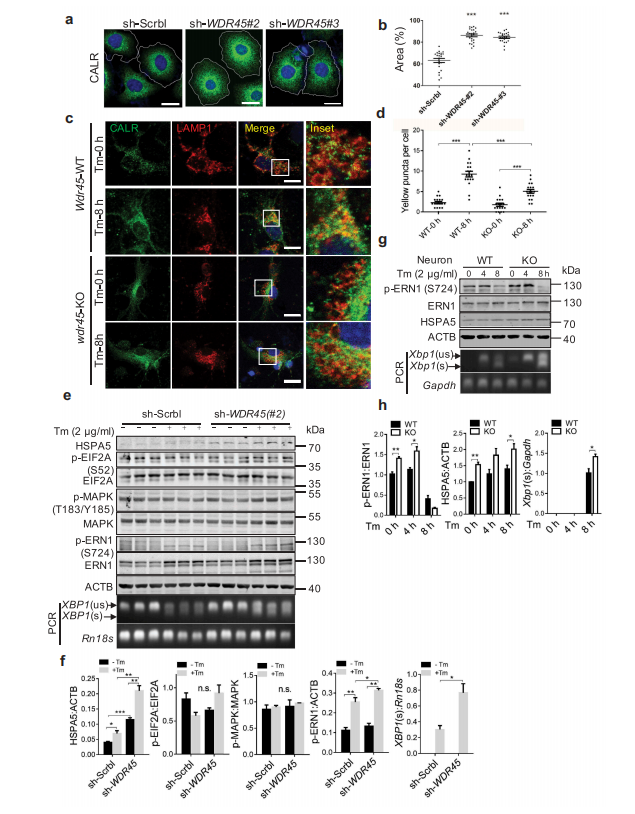
图6 WDR 45缺失细胞表现为ER应激增加,ER面积增加
6.WDR 45缺失导致ER应激后细胞死亡增加
接下来的实验结果表明,在WDR45缺失的原代神经元中,ER标记的Calr水平升高,自噬受体SQSTM1积累,LC3-II:LC3-I比率降低。作者利用western blot和免疫组化的方法对小鼠脑中以SQSTM1积累为特征的自噬缺陷和LC3-II:LC3-I水平降低,以HSPA5,DDIT3 / CHOP为特征的ER应激增加以及ERN1 / IRE1途径的激活,以及最终增加的细胞凋亡进行了重新描述。内质网应激诱导的细胞凋亡以CASP12的断裂为特征,在16个月龄时,作者还发现WDR45 KO小鼠PFC区CASP12的断裂增加。但在脑组织中UPR通路的激活存在差异。KO小鼠EIF2A磷酸化增加,提示EIF2AK3活化,ERN1/IRE1磷酸化和ATF6(N末端)水平变化不显著。在1月龄的KO小鼠中没有观察到内质网应激、未折叠蛋白反应和凋亡的缺陷,这与衰老是神经退行性变的一个重要危险因素的观点一致。
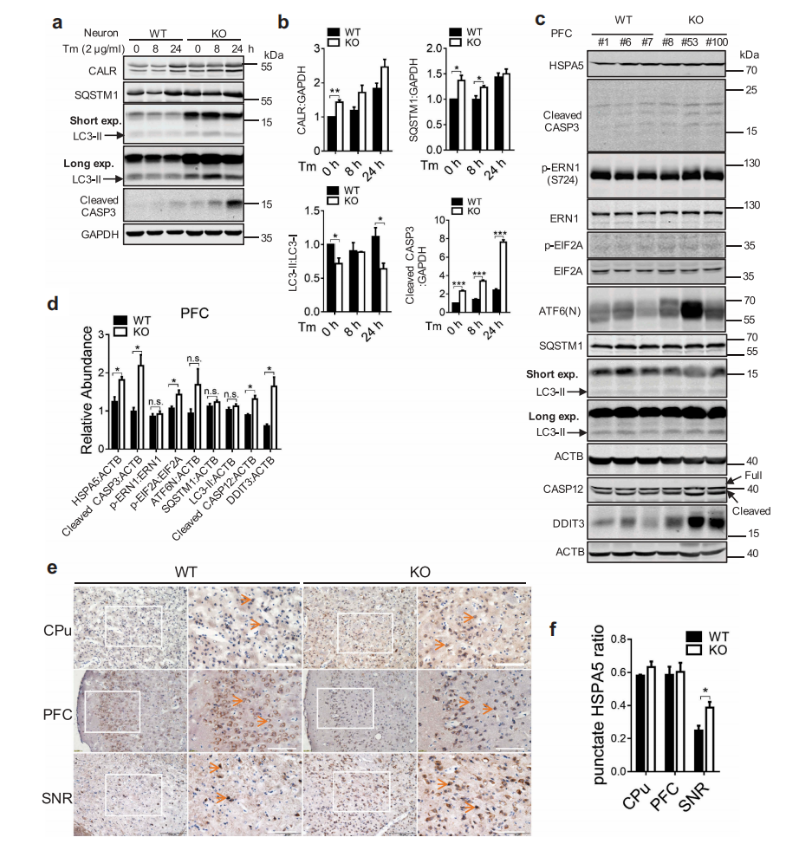
图7 WDR 45缺失导致ER应激后细胞死亡增加
7.激活自噬或抑制内质网应激挽救细胞凋亡
作者进一步猜想是否可以通过调节自噬和内质网应激水平来实现对细胞凋亡的药理挽救。作者使用雷帕霉素(mtor信号抑制剂)实验发现,雷帕霉素诱导自噬可能有助于预防或减轻神经退行性疾病的致病性蛋白聚集。此外,作者还使用了牛磺去氧胆酸(一种内质网应激抑制剂)进行实验。结果表明,雷帕霉素或牛磺去氧胆酸可减少WDR45缺失细胞中外源表达的ER蛋白SEC22B和SEC61B的积累。加重内质网雷帕霉素或牛磺去氧胆酸可减轻WDR45缺陷细胞的应激。同时雷帕霉素或牛磺去氧胆酸也降低了WDR45缺失细胞CASP3断裂的增加。流式细胞术标记细胞表面标志物ANXA5/ANNEXIN V和7-ADD的结果表明,雷帕霉素和牛磺去氧胆酸均可减轻tm诱导的细胞凋亡。最令人兴奋的是,在培养的原代神经元中,tm诱导的自噬活性在KO神经元中有缺陷。雷帕霉素和牛磺去氧胆酸可显著降低KO神经元CASP3的裂解。总之,这些数据提供了WDR45缺失导致自噬缺陷与内质网质量控制之间的完整联系;并提示β-螺旋桨蛋白相关神经变性病的潜在机制。
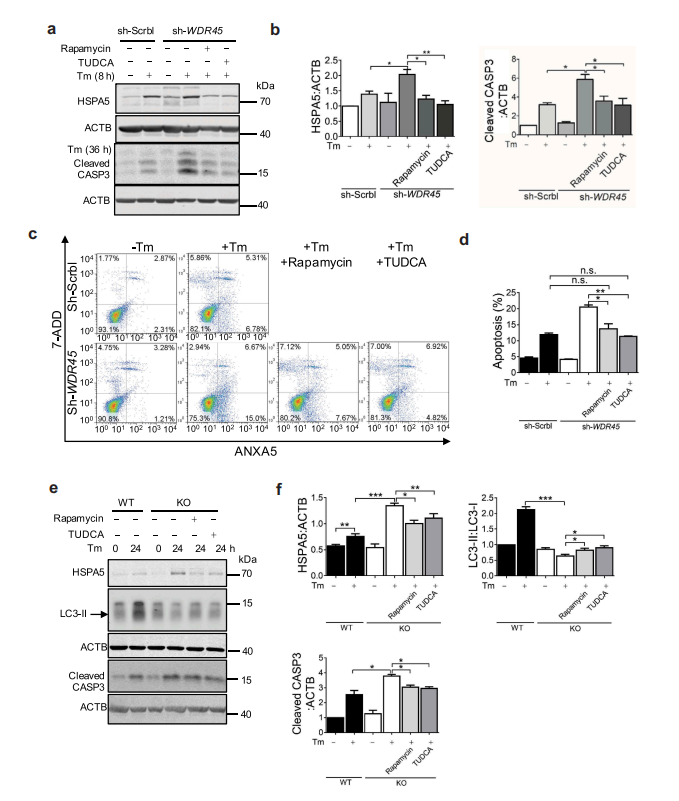
图8 激活自噬或抑制UPR可挽救异常蛋白质加工并增加WDR45缺失细胞的细胞死亡
小结
该研究利用CRISPAR-Cas9构建了WDR45 基因敲除小鼠,然后利用RT-PCR、PRM分别从基因水平和蛋白水平验证WDR45的敲除效果。一系列动物行为学实验,包括morris水迷宫实验、8臂迷宫试验、条件性恐惧实验、旋转实验等结果显示WDR45基因敲除小鼠具有认知障碍。然后作者利用3 x 10标TMT实验从分子层面研究了小鼠大脑组织的定量蛋白质组学,发现在基因敲除小鼠中的积累了大量的内质网(ER)蛋白。而后作者从细胞水平上验证发现WDR45缺乏可引起内质网蛋白积累,从而导致内质网应激增加和内质网质量控制受损。未折叠蛋白反应通过ERN1/IRE1或EIF2AK3/PERK途径升高,最终导致神经元凋亡。通过MTOR抑制内质网应激或激活自噬可减少细胞死亡。因此,WDR45的缺失会削弱神经元的宏自噬机制,并导致细胞器自噬的损伤,提供了对BPAN病因的机制性理解和治疗这种遗传性疾病的潜在治疗策略。